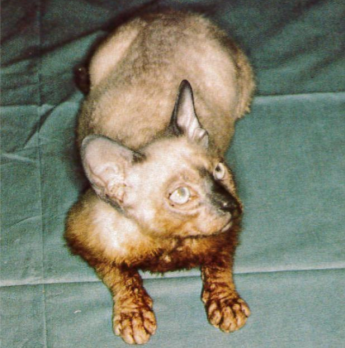
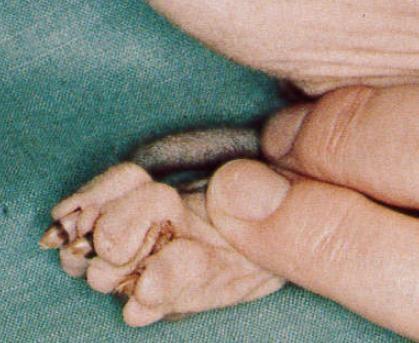
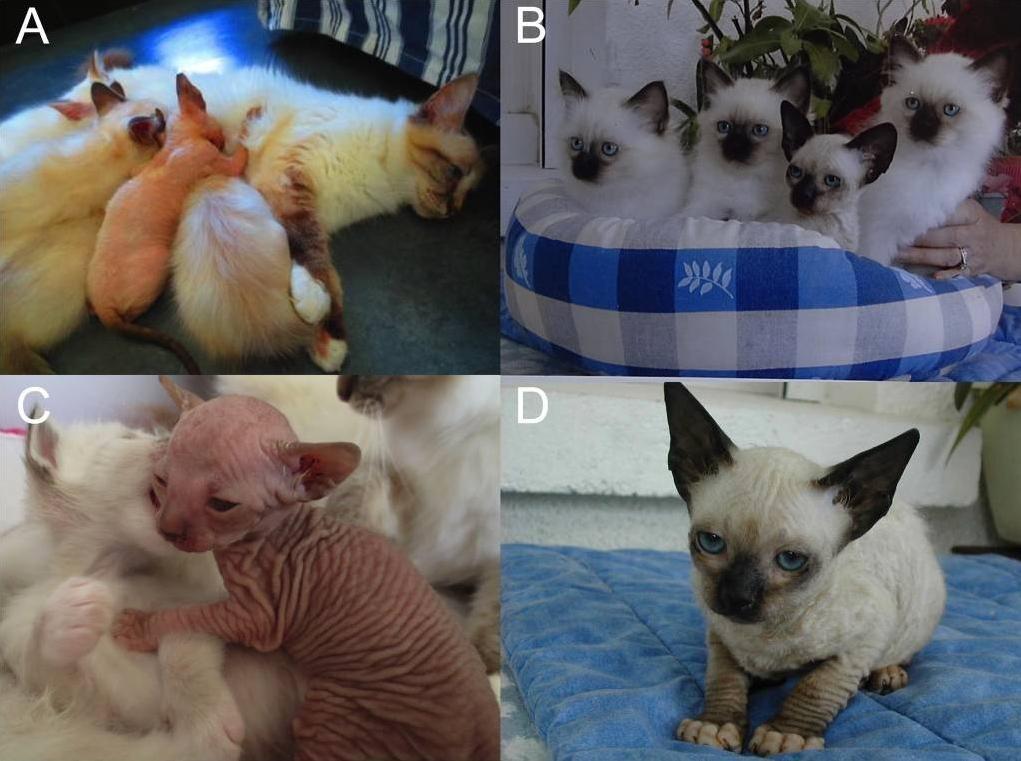
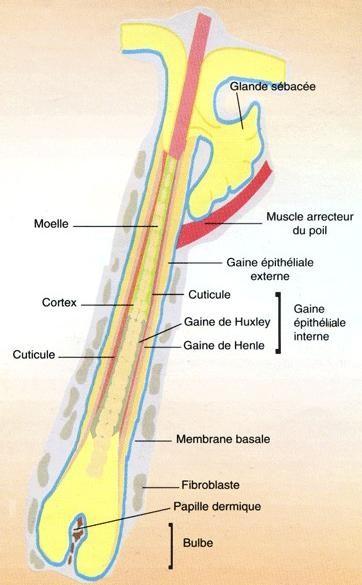
1. Chez le chat Sacré de Birmanie
Le gène FOXN1 du chat sous sa forme sauvage a été localisé sur le chromosome E1. Il présente 1944 paires de bases (pb) donnant une protéine de 647 acides aminés pour un poids moléculaire de 68 563,57 Dalton (« Ensembl », 2015). La protéine FOXN1 du chat présente un très fort degré d’homologie avec celle de la souris avec plus de 87 % de similarité, cette homologie était maximale au niveau du domaine Forkhead (99 %) et du domaine d’activation en C-terminal (98 %).
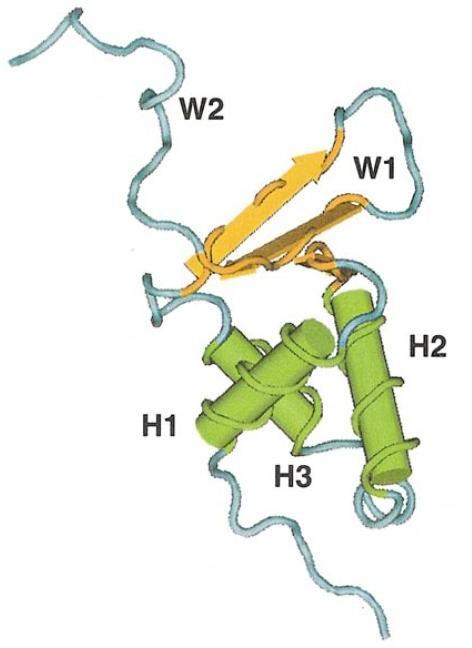
Il a été mis en évidence que la délétion de 4 pb identifiée au sein de l’exon 6 du gène FOXN1 des chats Sacré de Birmanie porteurs de la mutation de nudité provoquait un changement dans le cadre de lecture et une perte de la structure du domaine Forkhead. Cette perte de structure se traduisait par l’absence d’un feuillet β et de la boucle W2 du domaine FHD et était responsable de la perte de fonction du domaine en raison de l’incapacité à se lier à l’ADN.
De plus, l’apparition d’un codon stop prématuré entraînait la formation d’une protéine amputée d’une partie du domaine d’activation C-terminal, seuls 6 acides aminés sur 54 étaient conservés (identiques à la séquence normale) dans la séquence et les 15 derniers acides aminés étaient absents. En l’absence de ses domaines fonctionnels (domaine de liaison à l’ADN et domaine d’activation en C-terminal) la protéine FOXN1 ne pouvait remplir son rôle (Abitbol et al., 2015).
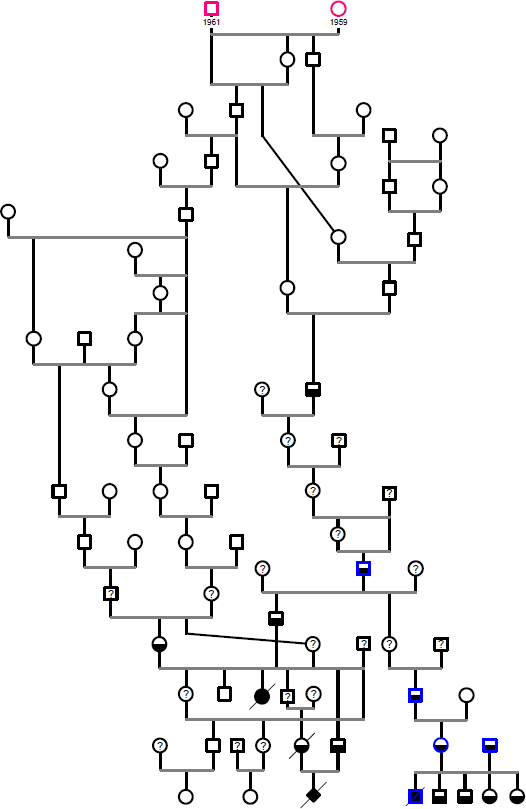
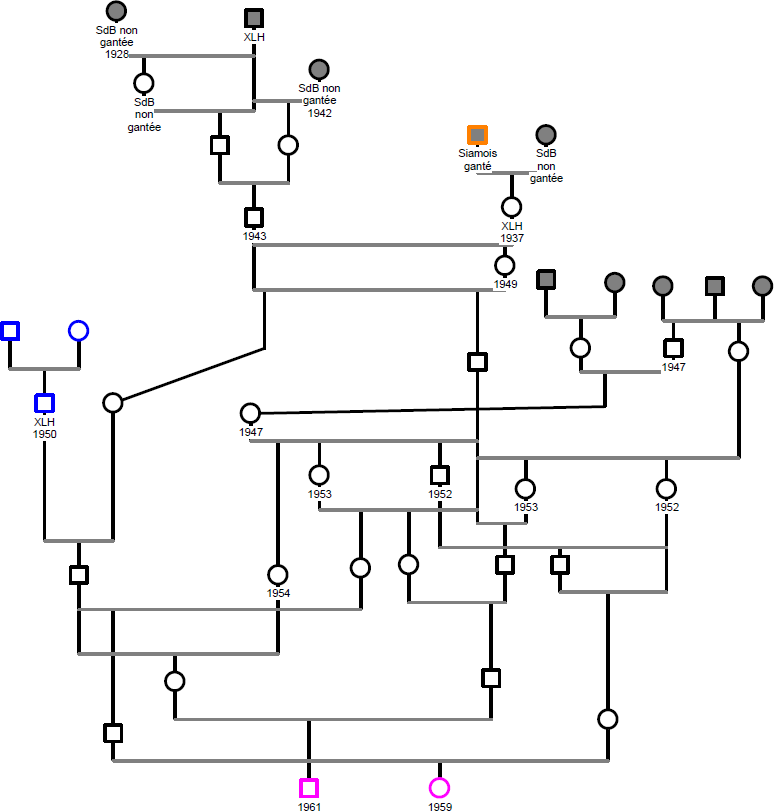
La mutation a été recherchée chez 215 chats de quinze autres races que Birman et n’a pas été retrouvée, mais elle était présente chez 3,2% des 126 Birmans recrutés dans l’étude. La délétion n’ayant pas été observée chez les races utilisées pour créer la race Sacré de Birmanie (Persan, Siamois et Orientaux), il était probable qu’elle soit apparue entre les années 1920 et 1977, après la création de la race Birman (Abitbol et al., 2015).
Un test génétique est proposé par le laboratoire Antagene depuis la découverte de la mutation causale du syndrome de nudité du Sacré de Birmanie. Ce test permet aux éleveurs de dépister les individus porteurs de la mutation et de réaliser des accouplements raisonnés en cas de reproducteurs hétérozygotes. Réalisé par le vétérinaire de manière non-invasive à l’aide d’un écouvillon buccal, le génotypage a son résultat connu sous quelques jours ( Antagene, 2015).
2. Chez les animaux de laboratoire
Le phénotype nude a également été retrouvé chez le rat : ces animaux présentaient les mêmes caractéristiques phénotypiques que les souris nudes à savoir une nudité et une immunodéficience sévère. Le gène responsable de ce phénotype chez le rat a été localisé sur le chromosome 10 et la région présentait une forte homologie en terme de séquence et d’organisation avec la région du locus nude de la souris (Cash et al., 1993).
Une mutation non-sens a été mise en évidence à la 1429 pb du gène rnu (rat-nude = FOXN1) chez le rat, cette mutation aboutissait à la formation d’une protéine non fonctionnelle (Segre et al., 1995).
3. Chez l’homme
Le syndrome de nudité de la souris a longtemps été comparé avec différents cas d’aplasie thymique chez l’homme, en particulier avec le syndrome de DiGiorge. Celui-ci provoque une aplasie totale du thymus mais également des autres composants issus du troisième arc branchial dont les parathyroïdes, la thyroïde et la mandibule, qui ne sont pas affectés chez les souris nudes (De Sousa et al., 1969 ; Pantelouris et Hair, 1970).
Un syndrome similaire au syndrome nude a été observé dans un village isolé d’Italie, Acerno. Les deux sœurs étudiées présentaient une alopécie congénitale et une dystrophie des ongles associées à une immunodéficience sévère. De plus aucun thymus n’a été observé sur les radiographies réalisées. La plus âgée est décédée prématurément à l’âge de 12 mois des suites d’infections récurrentes et à l’âge de 5 mois la plus jeune a reçu une greffe de moelle osseuse permettant une restauration complète des fonctions immunitaires mais sans modification de son aspect physique (Pignata et al., 1996).
De l’ADN a été prélevé sur des membres de la famille des deux sœurs et l’analyse du gène FOXN1 sur le chromosome 17 a mis en évidence une mutation non-sens en position 255 dans la protéine, au niveau de l’exon 5 du gène (R255X) conduisant à une protéine non fonctionnelle (Frank et al., 1999). Une étude réalisée en 2004 au sein du village d’Acerno a mis en évidence trois haplotypes portant la mutation et a révélé que 6,52 % de la population était porteuse à l’état hétérozygote de la mutation. Un couple d’ancêtres communs du début du 19ème siècle a été identifié (Adriani et al., 2004). Une campagne de consultations de génétique avec diagnostic pré-natal a été mise en place au sein du village en raison de la gravité du syndrome. En effet un cas de malformation sévère du tube neural a été observé chez un fœtus portant la mutation R255X, suggérant que le phénotype nude chez l’homme serait plus grave que celui de la souris (Amorosi et al., 2008).